Using GROMACS with chemlab¶
GROMACS is one of the most used packages for molecular simulations, chemlab can provide a modern and intuitive interface to generate input and analyze the output of GROMACS calculations. To illustrate the concepts we’ll perform a very simple simulation of liquid water.
Installing GROMACS¶
This depends on the system you’re using but I believe that GROMACS is already packaged for most linux distributions and also for other operating systems.
In Ubuntu:
$ sudo apt-get install gromacs
What GROMACS needs¶
In order to run a minimum simulation GROMACS requires to know some basic properties of the system we intend to simulate. This boils down to basically 3 ingredients:
- The starting composition and configuration of our system. This is provided by a ”.gro” file that contains the atom and molecule types, and their position in space.
- Information about the connectivity and interactions between our particles. This is called topology file and it is provided by writing a ”.top” file.
- Simulation method. This will require us to give parameters on how we want to make the system evolve. This is provided by an ”.mdp” file.
chemlab can help us to build any system that we want and we’ll use it to write a ”.gro” file. Then we will use chemlab to visualize and analyze the result of the GROMACS simulation.
Crafting a box of water¶
There are many ways to generate a box of water, in our example we will place 512 water molecules in a cubic grid. The advantages of doing that is the simplicity of the approach and the fact that we are naturally avoid any overlap between adiacent molecules.
To generate such a box we will:
- Create a template water
Molecule; - Translate this molecule on the grid points
- Add the molecule to a preinitialized
System.
import numpy as np
from chemlab.core import Atom, Molecule, System
from chemlab.graphics import display_system
# Spacing between two grid points
spacing = 0.3
# an 8x8x8 grid, for a total of 512 points
grid_size = (8, 8, 8)
# Preallocate the system
# 512 molecules, and 512*3 atoms
s = System.empty(512, 512*3)
# Water template, it contains export informations for gromacs
# more about export later...
water_tmp = Molecule([Atom('O', [0.0, 0.0, 0.0], export={'grotype': 'OW'}),
Atom('H', [0.1, 0.0, 0.0], export={'grotype': 'HW1'}),
Atom('H', [-0.03333, 0.09428, 0.0], export={'grotype':'HW2'})],
export={'groname': 'SOL'})
for a in range(grid_size[0]):
for b in range(grid_size[1]):
for c in range(grid_size[2]):
grid_point = np.array([a,b,c]) * spacing # array operation
water_tmp.move_to(grid_point)
s.add(water_tmp)
# Adjust boxsize for periodic boundary conditions
s.box_vectors = np.eye(3) * (8 * spacing)
# Visualize to verify that the system was setup correctly
display_system(s)
If you run this, it will display the following window:
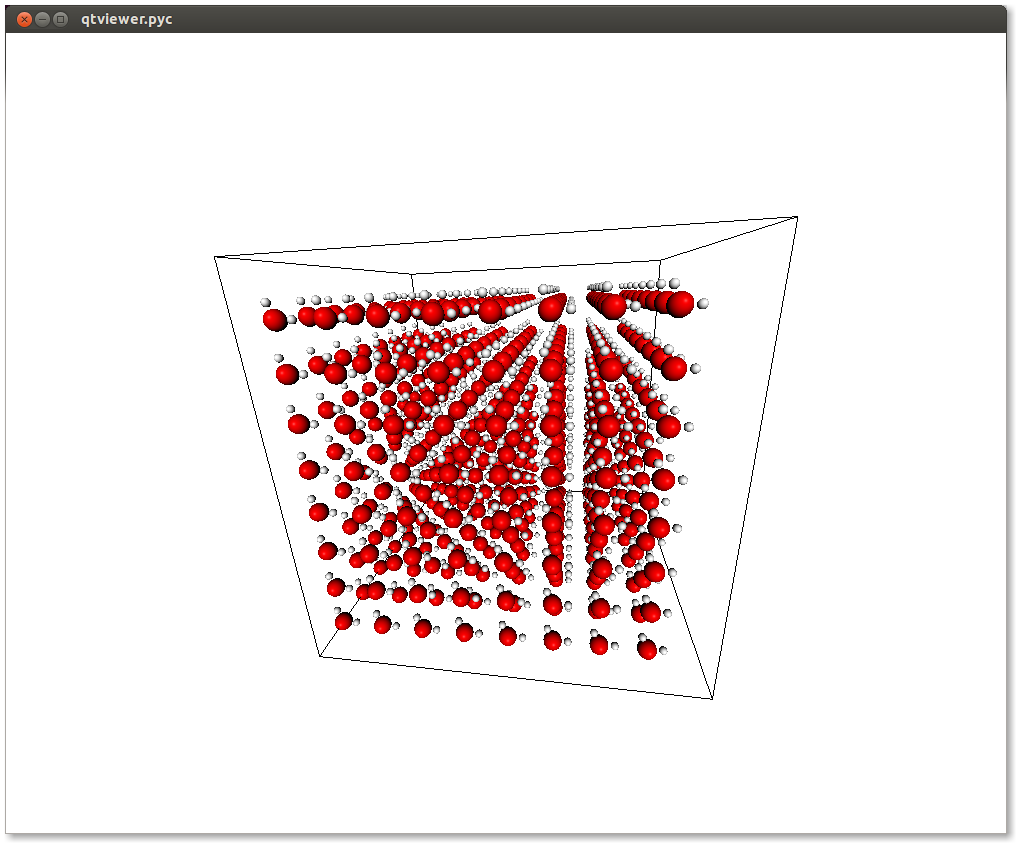
Awesome! Now we can write the ”.gro” file. Notice that when we defined our water molecule we had to pass an export dictionary to the atoms and molecules. The export mechanism is the way used by chemlab to handle all the variety of different file formats.
In this specific case, gromacs defines its own atom and molecule names in the ”.top” file and then matches those to the ”.gro” file to infer the bonds and interactions.
TODO Add picture of the export dictionary
How do we write the .gro file? Since we’ve already setup our export information, this is an one-liner:
from chemlab.io import datafile
datafile("start.gro", "w").write("system", s)
.top and .mdp files¶
I’ll give you directly the gromacs input files to do an NPT simulation of water, just create those files in your working directory:
topol.top
; We simply import ready-made definitions for the molecule type
; SOL and the atom types OW, HW1 and HW2
#include "ffoplsaa.itp"
#include "spce.itp"
[ system ]
Simple box of water
[ molecules ]
SOL 512
run.mdp
integrator = md
dt = 0.001
nsteps = 200000
nstxtcout = 100
rlist = 0.9
coulombtype = pme
rcoulomb = 0.9
rvdw = 0.9
dispcorr = enerpres
tcoupl = v-rescale
tc-grps = System
ref_t = 300
tau_t = 0.1
pcoupl = berendsen
compressibility = 4.5e-5
ref_p = 1.0
gen_vel = yes
gen_temp = 300
constraints = all-bonds
Running the simulation¶
To run the simulation with gromacs we have to do two steps:
Generate a parameter input, this will check that our input make sense before running the simulation:
grompp_d -f run.mdp -c start.gro -p topol.top
This will generate a bunch of files in your working directory.
Now we run the simulation, in the meantime, go grab coffee:
mdrun_d -v
This will take a while depending on your machine. If you are not a coffee drinker, don’t worry, you can stop the simulation by pressing Ctrl-C. The good news is that chemlab can read files from partial runs!
Viewing the results, the command-line way¶
To quickly preview trajectories and system energies you can use the script chemlab included in the distribution in scripts/chemlab.
GROMACS can store the trajectory (in the form of atomic coordinates) in the .xtc file. To play the trajectory you can use the command:
$ chemlab view start.gro --traj traj.xtc
Note
the nstxtcout = 100 option in the mdp file sets the
output frequency in the xtc file
You may also be interested to look at some other properties, such as the potential energy, pressure, temperature and density. This information is written by GROMACS in the ”.edr” file. You can use the chemlab script to view that:
$ chemlab gromacs energy ener.edr -e Pressure
$ chemlab gromacs energy ener.edr -e Temperature
$ chemlab gromacs energy ener.edr -e Potential
$ chemlab gromacs energy ener.edr -e Density
Warning
The chemlab gromacs command is a work in progress, the syntax may change in the future.
It is also possible to view and get the results by directly reading
the files and have direct access to the xtc coordinates and the energy
stored in the edr files. Take a look at the reference for
chemlab.io.handlers.XtcIO and
chemlab.io.handlers.EdrIO.
The tutorial is over, if you have any problem or want to know more, just drop an email on the mailing list python-chemlab@googlegroups.com or file an issue on github https://github.com/chemlab/chemlab/issues